


What next for COVID evolution?
Untangling the mysteries of antigenic turnover

Late 2020 ended with a nasty surprise. Or, rather, two nasty surprises: the variant that would become Alpha in the UK, and the Beta variant identified in South Africa. Despite the November 2020 announcement of highly effective vaccines, it was clear the challenges of COVID were not yet over.
The emergence of new variants would continue: Gamma, Mu, Delta, Omicron, BA.2, BA.4, BA.5… Which raises the question of how to interpret the evolutionary dynamics of SARS-CoV-2 viruses. What should we look for in the data? What can we learn from other viruses? And where might things being heading next?

Mapping the virus family tree
We can visualise the genetic relationship between different viruses by plotting them as a ‘phylogenetic tree’. There are a few different methods for doing this – each with their own assumptions and caveats – but the underlying principle is that viruses with a nearby common ancestor in the tree are more related than those with a distant common ancestor (much like human family trees).

As an example, here’s what the genetic relationships of SARS-CoV-2 variants looked like in March 2022, with each dot showing a sequenced virus and the lines showing the estimated phylogenetic tree from the great Nextstrain resource:
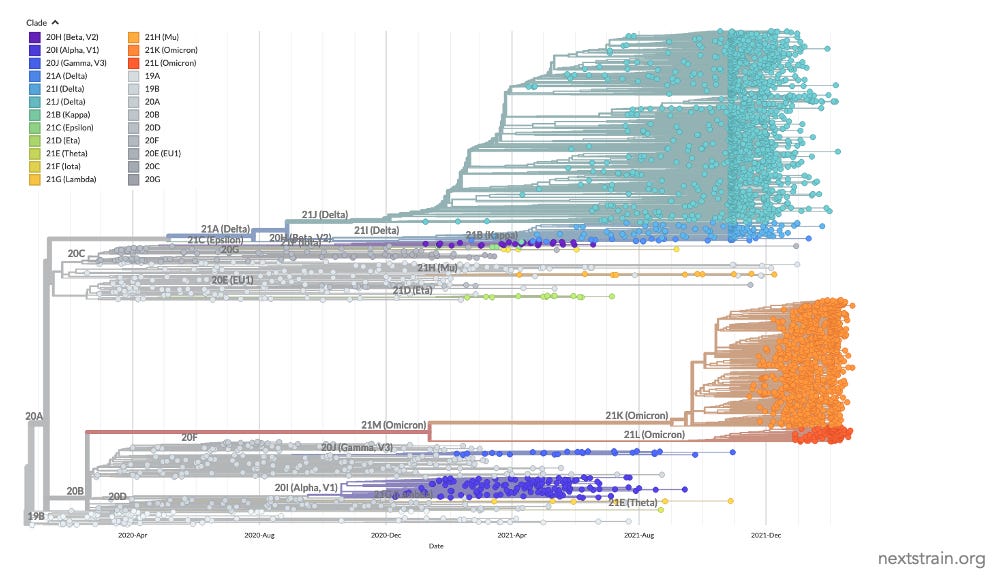
We can simplify the above plot by instead focusing on the ‘clade’ groupings for major variants:

You’ll notice that there was a mixture of more closely related variants (e.g. the original BA.1 Omicron variant and BA.2), alongside others that were distant cousins (e.g. the Delta variant that overtook Alpha in the UK and other countries in spring 2021). But how should we think about such patterns in real-time? It can help to look at some viruses that have been evolving for much longer.
Antigenic turnover
For infections like seasonal coronaviruses and influenza, we see a pattern of ‘antigenic turnover’ over time. Circulating viruses give rise to new variants that escape prior immunity against infection, immunity builds against these new variants, then these in turn spawn new variants.
When infections evolve to escape immunity, we typically end up with an phylogenetic tree that looks like a lopsided ladder as new variants sequentially replace their ‘parent’ variant lineages:

For example, here are the trees for seasonal coronavirus 229E (from Eguia et al) and seasonal flu virus A/H3N2 (adapted from Nextstrain):

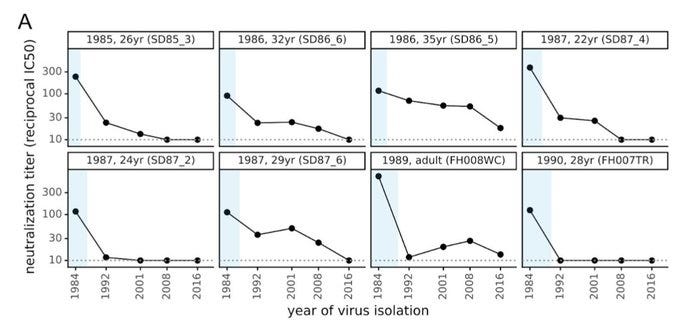
We can also see this signal of antigenic turnover in antibody data. Rachel Eguia and colleagues found that antibodies in samples taken at a point in the past (shown by blue boxes) were progressively less effective against more recent seasonal coronaviruses:
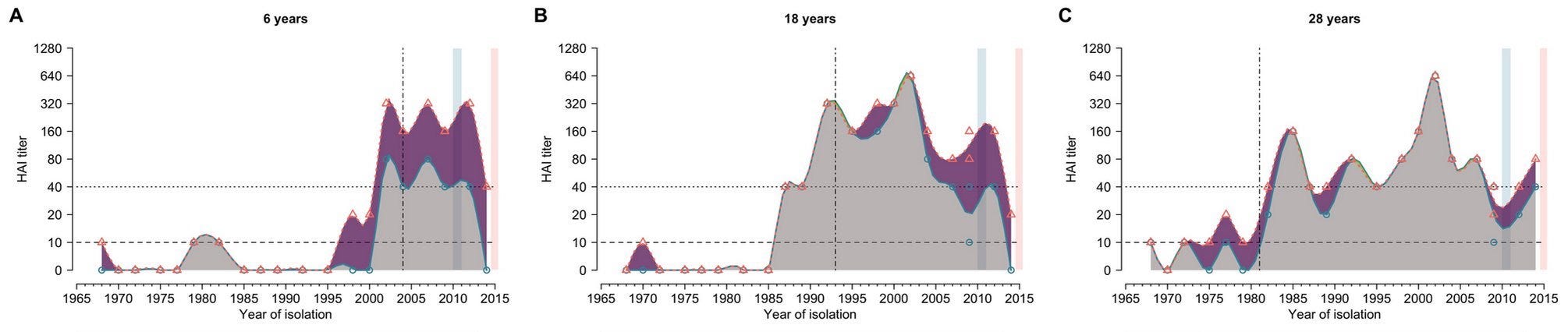
Similarly, Bingyi Yang et al showed that seasonal influenza antibody responses are less effective against strains that circulated before individuals were born (indicated by vertical dash lines):
We can also see evidence of adaptive evolution by looking at mutations in the so-called S1 region of the spike protein for seasonal coronaviruses (below shows analysis of OC43 and 229E by Kathryn Kistler and co). Because this is the part of the virus that antibodies target, we’d expect to see most change here if the viruses were undergoing antigenic drift:

Which brings us back to the phylogenetic tree of SARS-CoV-2 in spring 2022. Omicron hasn’t emerged from the Delta lineage and Delta didn’t emerge from the Alpha lineage. Given that we had to go so far back to find a common ancestor between Omicron and other circulating variants, one hypothesis is therefore that it emerged in an individual with a chronic infection.
But BA.2 was similar to BA.1, and soon BA.4 and BA.5 would be identified in South Africa. Were these early signals of a more familiar pattern of antigenic evolution? After the emergence of the 2009 influenza H1N1 pandemic, for example, the virus eventually settled into a pattern of gradual turnover, still causing epidemics today. Here’s the resulting phylogenetic tree for H1N1:
If we catch up on the most recent SARS-CoV-2 variants in Nextstrain (shown below), we can see more structure appearing, with the recent XBB lineages that emerged from among the BA.2 and BA.4 and BA.5 lineages, which in turn had replaced BA.1. This report by Tom Peacock and co from November 2022 has more detailed discussion of some of the changes behind these patterns.

Last week, the WHO declared EG.5 – a descendent of an XBB lineage – a ‘variant of interest’ (VOI) given its recent growth, alongside two other existing VOIs: XBB.15 and XBB.1.16. The accompanying report concluded that ‘due to its growth advantage and immune escape characteristics, EG.5 may cause a rise in case incidence and become dominant in some countries or even globally.’
But SARS-CoV-2 might not be settling into a neat pattern of antigenic drift just yet. In recent days, three SARS-CoV-2 viruses descended from BA.2 have been identified in Denmark and Israel with a large number of mutations. In genetic terms, they are a remarkably distant cousin of Omicron, just as Omicron was a remarkably distant cousin of the original SARS-CoV-2 virus. (As an aside, early details of this virus were posted on Twitter, illustrating the continuing value of having a social media hub for scientific discussion.)
So, what do these mutations mean? Will the variant spread widely, will it affect patients differently, or is it a mere curiosity that will fade away into obscurity?
From genotype to phenotype
In late 2022, the XBB Omicron subvariant led to tens of thousands of cases in Singapore, despite more than 90% of the population being vaccinated. Early lab data suggested that subvariants like XBB, XBB.1.5, and BQ.1.1 could evade prior immunity, which would imply potential for widespread reinfection globally.
However, when Ed Carr and colleagues looked at antibody responses among individuals in a UK cohort who’d lived through multiple large COVID waves, they found individuals who’d had a bivalent vaccine (i.e. containing BA.1) or a prior Omicron infection generally had strong neutralising antibodies against the new variants. As a result, the winter 2022-23 UK wave was not as severe as some had feared.
The timing and extent of past infection can make a big difference to future SARS-CoV-2 epidemics. When the original Omicron variant took off in late 2021, the impact on South Africa was less severe than in some earlier waves, despite only 1 in 4 people having been vaccinated. In contrast, when Omicron hit Hong Kong – with its overall vaccine coverage just above 60% – death rates rocketed. As well as the age differences in the populations, South Africa had seen a lot more SARS-COV-2 infection prior to the emergence of Omicron.

It’s not just variation in population exposure that makes it hard to predict the impact of new variants. In the world of influenza, it’s been a long standing challenge to predict which viruses will become dominant in future, and hence which ones to put in next season’s vaccine. As well as genotype (i.e. genetic profile of a virus) it’s also about phenotype (i.e. what happens during a resulting infection). More mutations doesn’t necessarily mean less immunity.
Take the below plot from Trevor Bedford and colleagues, which shows the relationship between two ways of measuring the ‘distance’ separating influenza viruses A and B: 1) similarity in a phylogenetic tree (‘tree distance’), and 2) strength of antibody response against virus B after infection with virus A (‘antigenic distance’):
It’s therefore not enough to only at the number of genetic mutations; we need to find ways to combine datasets to investigate what they might mean. For example, there’s evidence that linking data on genetic mutation with phenotype insights – whether from immune experiments or growth rates in human population – can lead to better forecasting performance for influenza.
In the case of SARS-CoV-2, Bernadeta Dadonaite and colleagues have outlined a ‘deep mutational scanning’ approach, building libraries that document how different combinations of mutations affect two viral phenotype properties: 1) how well antibodies recognise the virus and 2) how well the virus can infect cells in the absence of antibodies.
Earlier today, Jesse Bloom shared a summary from a mutational scanning of the new virus identified in Denmark and Israel. Several mutations were of interest from an immunological point of view, and the analysis reiterated just how large an evolutionary jump this variant has made. However, Bloom notes: ‘to be successful, variant would need to combine this antigenic advantage with inherent transmissibility at least close to current XBB variants.’
Despite progress in understanding the relationship between genotype and phenotype, predicting what a variant will do in the real world remains a major scientific challenge. Even if a variant picks up mutations that give it an advantage over others in terms of evading immunity – and we can predict the effect of those mutations at the individual level – it would also need to spread from person-to-person. And even if we could accurately predict this person-to-person transmission potential, randomness in the early transmission process may cause it to stutter to extinction before causing a major epidemic.
Where next?
When considering what the long-term evolution of SARS-CoV-2 could look like, I think seasonal coronaviruses and influenza remain useful prior indicators. But we should also remember this process of sequential turnover hasn’t been smooth so far. Like Omicron BA.1 and Delta before it, the next variant may break the recent pattern of evolution.
This shows the importance of having good ongoing surveillance of SARS-CoV-2 genetic sequences globally, optimised to make best use of resources. It also shows the importance of capacity to rapidly test the characteristics of these viruses, and relationship to other circulating variants. And the need for ongoing cohort studies, so we can understand the role of prior infection and vaccination in shaping what will happen next. What’s more, we’ll need quantitative methods to link all these datasets together, uncovering key characteristics and identifying ways to make better predictions.
There may be another nasty SARS-CoV-2 surprise on the way. Perhaps sooner, perhaps later. But let’s make sure we’re in a position to find out about it – and what it really means – as quickly as we can.
Update 18th Aug: There have now also been reports of the new BA.2 descendant variant (now designated BA.2.86) in Michigan and London, as well as existing sequences identified in Denmark and Israel. It’s notable that all of these countries do a lot of genetic sequencing relative to others, which suggests that there are likely to be other undetected BA.2.86 cases globally.
I wonder if anyone can answer a question that has been on my mind for quite a while. If I understand correctly, it is essentially the specific shape of the spike protein that allows it to bind to and infect human cells. But isn't it also the specific shape of the spike protein that allows neutralizing antibodies to bind to and inactivate the virus? At some point shouldn't viral mutation reach a point of diminishing returns, in that a mutation that decreases the effect of neutralizing antibodies also decreases the ability of the virus to infect cells?